How Do Mutated Hemoglobin Genes Cause Sickle Cell Disease?
Reviewed by: HU Medical Review Board | Last reviewed: January 2021 | Last updated: June 2021
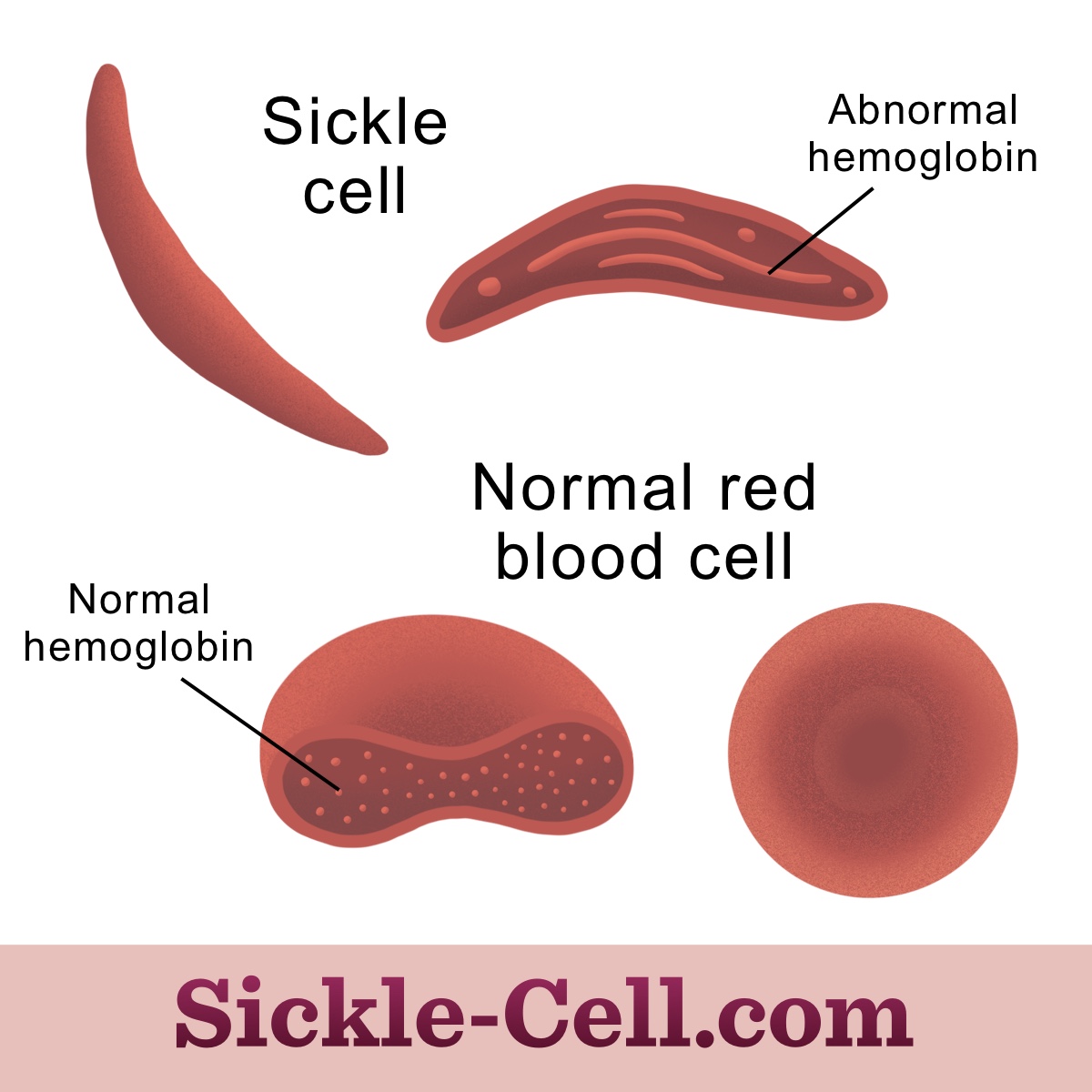
Sickle cell disease (SCD) is a red blood cell disorder caused by abnormal hemoglobin. Red blood cells (RBCs) are the transport system our bodies use to deliver oxygen around the body. They are rich in a protein called hemoglobin, which binds oxygen in the lungs and releases it around the body.
Normal RBCs are flexible discs that can bend to move through small blood vessels. People with SCD have rigid, sickle-shaped RBCs because of abnormal hemoglobin. Sickle cells can block blood flow and burst apart too soon. This may lead to acute pain crises, anemia, and other complications.
How do hemoglobin and red blood cells normally function?
Hemoglobin and RBCs must function properly to quickly deliver a lot of oxygen from the lungs to the rest of the body. Hemoglobin is an iron-containing protein in RBCs that binds and releases oxygen. It is responsible for the red color of blood.1
The most common form of normal hemoglobin is called hemoglobin A. It is made up of 4 smaller proteins: 2 “alpha” subunits and 2 “beta” subunits. Each subunit contains a “heme” group, which is the iron-containing center that directly binds and releases 1 molecule of oxygen. This means each hemoglobin protein can transport 4 molecules of oxygen.2
Red blood cells are essentially sacs of hemoglobin. Each RBC contains about 270 million hemoglobin molecules.3 RBCs develop in the bone marrow and circulate for about 120 days before being degraded and recycled in the spleen.4
RBCs are shaped like disks with a high surface area so that oxygen has plenty of space to pass in and out of the cell. They are also very flexible so they can squeeze through tiny blood vessels. We have about 20 to 30 trillion RBCs at any time, which is about 70 percent of all of our cells.5,6
What happens to hemoglobin and red blood cells in sickle cell disease?
Hemoglobin is enriched in RBCs, so abnormal hemoglobin severely impacts how RBCs function. Several genes provide instructions for our body to make the hemoglobin subunits. SCD is caused by mutations in the HBB gene, which encodes the “beta” subunit of hemoglobin.2,7
The specific mutation on hemoglobin depends on the type of SCD. The most common mutation causes people to have sickle hemoglobin, or hemoglobin S. Sickle hemoglobin clumps together to form rigid strands within RBCs when oxygen levels are low. These strands change the way RBCs work and makes them shaped like rigid sickles.4,7,8
Figure 1: Normal hemoglobin compared to hemoglobin S
Thalassemia is similar to SCD because it is also caused by mutations in hemoglobin genes. However, thalassemia mutations do not cause hemoglobin to form rigid strands. Instead, they usually cause the body to produce lower amounts of hemoglobin subunits. The severity depends on the subunits affected and how much hemoglobin is produced.2
How do sickle red blood cells cause complications?
Sickle cells are rigid and stick to the walls of blood vessels. This can block blood flow and prevent oxygen from reaching parts of the body. When this happens, stroke or sudden attacks of pain can occur. These pain crises can strike almost anywhere in the body, but often occur in the:1,2
- Abdomen
- Arms
- Back
- Chest
- Legs
Pain crises can occur without any warning. They are the most common cause for people with SCD to seek hospital care.1,2
Sickle cells also rupture too soon because they cannot change their shape easily. They only circulate for about 10 to 20 days, while normal RBCs circulate for about 120 days. The body cannot produce new RBCs fast enough to keep up with this quick turnover. This means people with SCD often have a lower number of RBCs.4
This can lead to severe anemia and jaundice (yellowing of the skin/eyes). It can also cause the spleen to become enlarged as it traps RBCs that should be circulating in the bloodstream. Damage to the spleen lowers the body’s ability to fight mild infections.1
When sickle cells burst apart too soon, they release hemoglobin into the bloodstream. Free hemoglobin affects the way the body regulates blood pressure. Free heme can also cause inflammation.9