Sickle Cell Crisis
Reviewed by: HU Medical Review Board | Last reviewed: January 2021 | Last updated: June 2022
Sickle cell crisis is the most common complication of sickle cell disease (SCD). Acute episodes of pain can occur anywhere in the body and may lead people to seek hospital treatment.
Sickle cell crises happen when blood flow and oxygen delivery are blocked by sickle cells. Pain episodes may trigger other complications of SCD, and multiple episodes can cause long-term damage. Most pain events are managed at home, but severe pain must be treated in a hospital.
What is a sickle cell crisis?
The sickle cell crisis, also called acute pain crisis or vaso-occlusive crisis, is the most common reason that people with SCD go to the hospital. Episodes are sudden and unpredictable and may be triggered by different unknown risk factors.1,2
About 5 percent of people with SCD have 3 to 10 episodes of severe pain per year. The average rate is about 1 episode per year for people with sickle cell anemia and sickle beta zero thalassemia. The average is about 1 episode per 2 years for people with hemoglobin SC disease and sickle beta plus thalassemia.3
Sickle cell crises often lead to further complications of SCD. About half of reported cases of acute chest syndrome happen after a hospital stay for a pain crisis. Sometimes, sickle cell crises can result in acute failure of multiple organs or sudden unexpected death.4-6
The sickle cell crisis is different from other sudden complications, such as aplastic crisis and splenic sequestration crisis. These are complications that can cause severe anemia. The pain during a sickle cell crisis is different from chronic pain, which can be caused by other complications of SCD, such as avascular necrosis of joints.2,7
What causes pain crises?
People with SCD experience severe acute pain because of many factors. Sickle red blood cells block blood flow because they can stick to blood vessel walls. Also, they are not flexible enough to pass through small blood vessels. This can lead to hypoxia (low oxygen levels) and ischemia (reduced blood flow), which can damage many organs.8,9
Figure 1: Sickle cells can block blood vessels leading to hypoxia and ischemia.
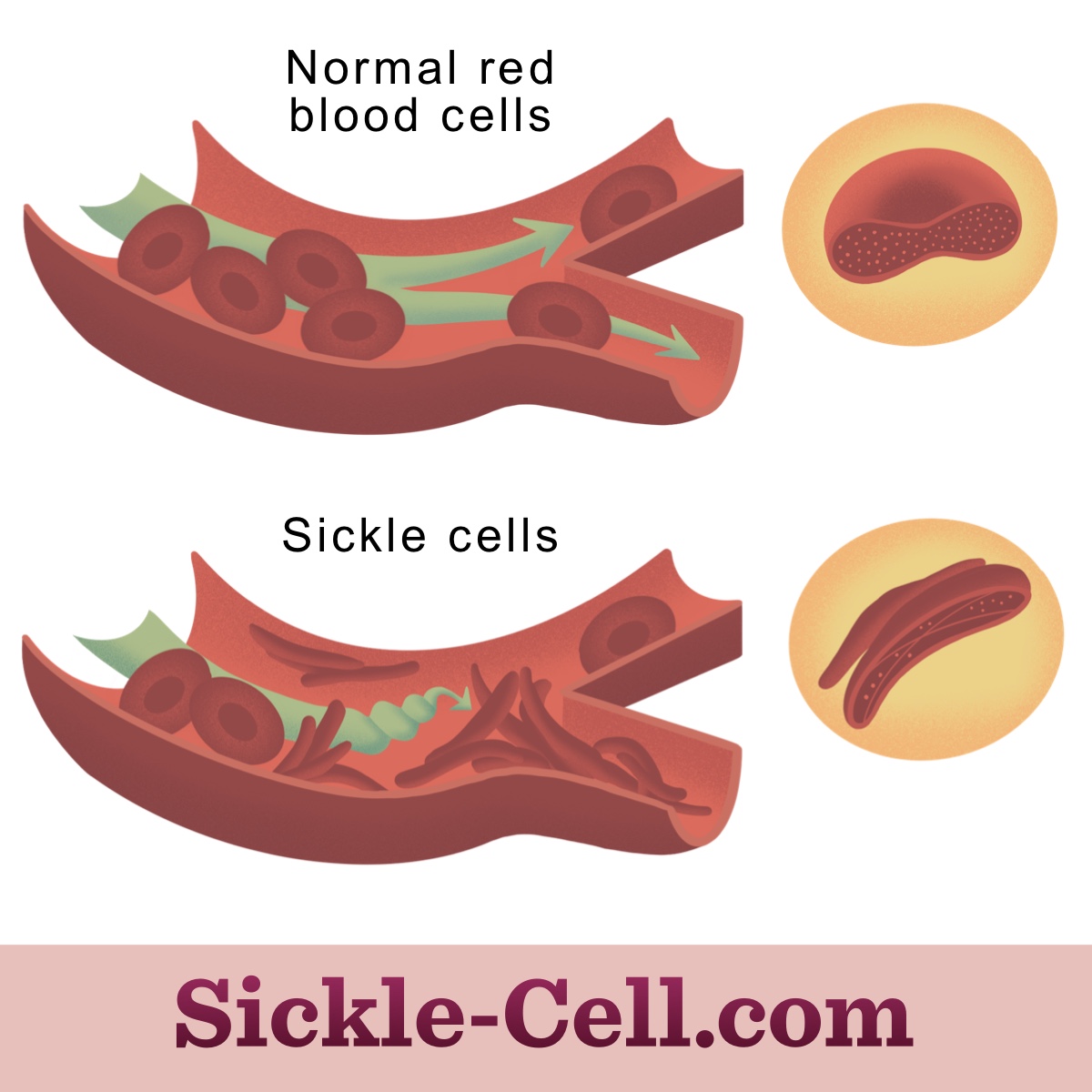
In addition, sickle cells tend to rupture quickly and easily. When they burst, they release molecules that can trigger immune cells and promote inflammation.8,9
Sickle cell crises are often triggered by inflammatory or environmental factors. These can include:10
- Dehydration
- High altitude
- Infection
- Stress
- Temperature changes
What are the symptoms?
A typical pain crisis is a sudden onset of pain in the lower back or in joints of the arms and legs. The pain may be:2
- In a specific area or move to multiple areas
- Continuous and throbbing
- A cause of groaning, crying, and twisting and turning to try to relieve pain in infants/children
- Described as sharp, pounding, dull, stabbing, cutting, or gnawing
Sickle cell crises seem to develop in specific phases. The first phase usually lasts 1 to 2 days. During this phase, people may experience numbness, prickling, and aches in a certain area of the body. Then, a phase of increasing pain in the area occurs. Anxiety, fear, and problems with ER personnel often occur during this phase. Maximum pain then lasts for several days before resolving.11
How is crisis prevented and treated?
Acute pain crises may be reduced by:12
- Avoiding extreme temperatures
- Avoiding high altitudes and exposure to low oxygen
- Drinking plenty of water
- Getting a yearly flu shot and staying current on vaccines
- Taking antibiotics to prevent infections
- Taking hydroxyurea
Even though acute pain crises are the most common complication of SCD, treatment needs to be improved. Most acute pain events are managed at home with non-steroidal anti-inflammatory drugs (NSAIDs) like aspirin or ibuprofen, or oral opioid pain medications. When this does not work, pain crises can be treated in a hospital with stronger oral and IV opioids.8
However, people with SCD often see a delay in treatment for acute pain because of discrimination. People with SCD, who are mostly Black, on average wait 25 to 50 percent longer than people without SCD who have similar levels of pain.13
Hospitalization may not be the end of the sickle cell crisis. About half of people with SCD who are admitted to a hospital for a painful episode are readmitted within a month. The major cause of readmission is sickle cell pain that was not controlled at home or in the ER.1